Синдром ретта у детей
Содержание:
- Схожие с синдромом Аарскога расстройства
- Причины возникновения
- Чем лечат синдром Туретта в Москве
- Лечение синдрома Ретта
- Решение проблемы на мировом уровне
- Лечение
- Симптомы синдрома Ретта
- Диагностика заболевания
- Что такое «синдром Ретта»?
- Причины появления
- Как обнаружить и подтвердить диагноз?
- Стадии болезни
- Симптомы синдрома Аарскога
Схожие с синдромом Аарскога расстройства
Симптомы следующих расстройств могут быть схожими с таковыми при синдроме Аарскога. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Нунана является относительно распространенным генетическим заболеванием, характеризующимся:
- низким ростом
- дисморфными чертами лица
- врожденным пороком сердца.
Расстройство характеризуется широким спектром симптомов и физических особенностей, которые сильно различаются по степени тяжести. У многих пораженных людей связанные нарушения включают:
- отличительный внешний вид лица;
- широкая или перепончатая шея;
- низкая задняя линия роста волос;
- типичная деформация груди
- низкий рост.
Характерные аномалии головы и лица (черепно-лицевой) области могут включать:
- широко расставленные глаза (глазной гипертелоризм);
- кожные складки, которые могут покрывать внутренние углы глаз (эпикантальные складки);
- опущение верхних век (птоз);
- маленькую челюсть (микрогнатия);
- подавленный носовой корень;
- короткий нос с широким основанием;
- низко посаженные назад повернутые уши.
Также обычно присутствуют отличительные пороки развития скелета, такие как аномалии грудины (грудины), искривление позвоночника (кифоз и/или сколиоз) и вальгусное колено. Многие дети с синдромом Нунана также имеют проблемы с сердцем, такие как обструкция правильного кровотока из нижней правой камеры сердца в легкие (стеноз легочных клапанов).
Дополнительные аномалии могут включать пороки развития определенных кровеносных и лимфатических сосудов, свертываемость крови и дефицит тромбоцитов, трудности в обучении или умеренную интеллектуальную неспособность, неспособность яичек опускаться в мошонку (крипторхизм) к первому году жизни у пораженных мальчиков и/или другие симптомы и признаки.
Синдром Нунана является генетически гетерогенным состоянием, которое может быть вызвано мутациями ряда генов PTPN11, KRAS, SOS1, RAF1, NRAS, RIT1 и SOS2.
Синдром Робинова — это редкое генетическое заболевание, наследуемое как доминантный и рецессивный признак и характеризующееся:
- невысоким ростом из-за задержек роста после рождения;
- отличительными аномалиями головы и лица;
- дополнительные пороки развития скелета;
- генитальные аномалии.
Черты лица у детей с синдромом Робинова напоминают черты лица восьминедельного плода; в медицинской литературе это состояние часто упоминается как «лицо плода».
Характерные черепно-лицевые особенности могут включать:
- аномально большую голову (макроцефалия) с выпуклым лбом;
- широко расставленные глаза;
- маленький вздернутый нос с ноздрями, расклешенными вперёд;
- затонувший (вдавленный) носовой мост.
Пороки развития скелета могут включать необычно короткие кости предплечья (брахимелия предплечья), аномально короткие пальцы рук и ног, постоянную фиксацию пятого пальца в согнутом положении (клинодактилия), необычно маленькие руки с широкими большими пальцами, порок развития ребра, сколиоз и/или недоразвитие одной стороны костей в средней (грудной) части позвоночного столба (гемивертебра).
Генитальные аномалии, связанные с синдромом Робинова, могут включать аномально маленький пенис (микропенис) и неспособность яичек опускаться в мошонку (крипторхизм) у пораженных мужчин и недоразвитие (гипоплазия) клитора и наружных удлиненных складок кожи с обеих сторон вагинального отверстия (половых губ) у пораженных женщин.
Диапазон и серьезность симптомов варьируются от случая к случаю.
Синдром Робинова является генетически гетерогенным состоянием, которое может быть вызвано мутациями в разных генах, таких как WNT5A, ROR2, DVL3 и DVL1.
Причины возникновения
Точные причины возникновения синдрома Ретта до сих пор остаются предметом дискуссий, поэтому можно привести в пример несколько наиболее распространенных теорий. Все они сейчас проверяются путем проведения различных исследований, и хорошо еще, что медики хотя бы установили отличие от слабоумия и теперь движутся в правильном направлении.
Чаще всего специалисты указывают на генное происхождение болезни: они говорят, что недуг вызван мутацией генов. Данное отклонение развивается до масштабов патологии еще на этапе вынашивания плода, поэтому последствия видны уже на ранних этапах жизни.

Указывается, что повышенное количество кровных связей в родословной наверняка влияет на вероятность развития синдрома Ретта, причем процент таких связей высок лишь по сравнению с нормой – 2,5% вместо 0,5%. В пользу такого фактора, как причины возникновения патологии, говорят и ярко выраженные очаги заболеваемости – обычно это небольшие, достаточно изолированные от мира деревушки, где половые связи между членами одной семьи в прошлом вполне вероятны.
Другая группа исследователей указывает на нарушения в Х-хромосоме – ломкость одного из участков ее короткого плеча.
Доказано, что иногда такая аномалия действительно наблюдается, но ученые пока не смогли обнаружить точный участок, отвечающий за развитие патологии, равно как и установить определенную закономерность, позволяющую утверждать стопроцентную взаимосвязь причины и следствия.
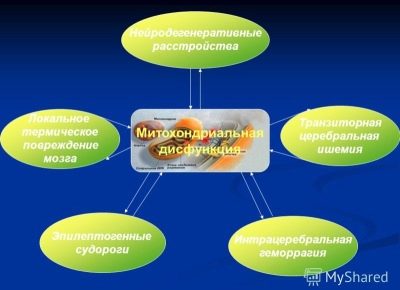
Существует и третья (менее известная) теория, согласно которой синдром Ретта может быть следствием метаболических аномалий – нарушенного обмена веществ, вызванного митохондриальной дисфункцией.
Отмечается, что у всех больных данным заболеванием отмечено повышенное содержание в крови двух кислот – пировиноградной и молочной, очевидны также патологии лимфоцитов и миоцитов. Впрочем, это пока только наблюдение, и специалисты сейчас не могут сказать, является ли это причиной возникновения синдрома Ретта или его следствием.
Чем лечат синдром Туретта в Москве
Все методы, которые используются при лечении подобных заболеваний, можно поделить на медикаментозные и немедикаментозные. Первые предполагают прием седативных препаратов, редко транквилизаторов. Если их и назначают, то короткими курсами и только после того, как врач выяснит, что это необходимо.

Немедикаментозные методы лечения синдрома Туретта:
- расслабляющий массаж – ручной и аппаратный;
- рефлексотерапия. Вариант выбирается в зависимости от ситуации. Такую терапию проводят при помощи рук, специальных игл, электрических импульсов. Смысл в том, чтобы воздействовать на определенные точки тела, которые помогут расслабиться, стабилизировать нервную систему;
- ароматерапия. Она направлена на отдых, успокоение, расслабление. После консультации с врачом ароматерапия доступна и в домашних условиях – это простой и часто действенный метод;
- лечебный сон (электросон). Благодаря импульсам тока через специальные аппараты такой сон нормализует работу мозга, ЦНС. Обычно он длится от 30 минут до 2 часов;
- психотерапия. Она помогает человеку избавиться от тревожности, сложных состояний, психологических травм, а также учит принимать свое состояние.
Важный момент при лечении синдрома Туретта – это правильная обстановка дома, среди близких друзей. Человек с таким отклонением должен получать поддержку, заботу, его необходимо оградить от насмешек и неадекватных реакций на тики.
Лечение синдрома Ретта
Чтобы выявить развитие патологии, врачи проводят диагностику, направленную на обнаружение изменений в головном мозге. Ребенка могут направить на:
- МРТ и КТ. Исследования проводят для того, чтобы определить участки атрофии головного мозга, если они имеют место быть.
- ПЭТ. Дает возможность выявить значительное снижение мозгового кровообращения.
- ЭЭГ. Дает возможность изучить электрическую активность мозга. Если наблюдается заторможенность фонового ритма, это может свидетельствовать о развитии патологии.
В случае подозрения на задержку в развитии, родители ребенка должны обратиться к неврологу. Зачастую синдром Ретта путают с детским аутизмом. Однако этологии отличаются друг от друга. Так, при синдроме Ретта наблюдается задержка роста, дыхательные нарушения и припадки. При раннем детском аутизме такие проблемы отсутствуют, однако возможно появление умственной отсталости до одного года. При синдроме Ретта такое явление не возникает. Различаются и движение, которое может совершать пациент.
Сегодня не существует лекарственных препаратов, позволяющих избавиться от патологии. Однако существует разработанный ряд методик, дающий возможность улучшить состояние пациента. Проведение медикаментозной терапии дает возможность снизить интенсивность проявления характерных симптомов. Чтобы контролировать эпилептические припадки, врач назначит приём Карбамазепина или Ламотриджина.
Дети, болеющие патологией, страдают проблемами со сном. Чтобы избавиться от симптомов, используют аналог гормона Мелатонин. Для того, чтобы улучшить мозговое кровообращение, применяют Пирацетам, Этирацетам.
Помимо медикаментозной терапии используются методы реабилитации. Родители больного ребенка должны посещать вместе с ним психолога и логопеда минимум один раз в неделю. Проводятся сеансы массажа и лечебной оздоровительной физкультуры. Действия направлены на укрепление мышц и увеличение их тонуса. Полезно проведение музыкотерапии.
Хорошие отзывы заслужил метод, который придумал французский отоларинголог Альфред Томатис. Цель метода состоит в повторном обучении ребенка процессу мышления. Это позволяет улучшить способности к изучению и освоению языков, а также увеличить творческий потенциал. Всё это положительно сказывается на социальном поведении ребёнка.
Дополнительно проводятся арт-терапии и дельфинотерапия. Вышеуказанные методы оказывают положительное влияние на психоэмоциональное состояние ребенка. Посещение остеопата также позволяет улучшить состояние больного. Прогресс наблюдается уже после нескольких сеансов. Пациенту может быть назначена иппотерапия и гидротерапия. Вышеуказанные методы оказывают химическое воздействие на тело ребёнка и укрепляют его мышцы. АВА-терапия дает возможность улучшить социальную адаптацию больных. Сложные развивающие действия разбиваются на мелкие части. В будущем они собираются в единый блок.
Решение проблемы на мировом уровне
После столь шокирующего открытия была создана специальная ассоциация синдрома Ретта. Это мировая организация пришла к выводу, что дети, пораженные недугом, нуждаются в реабилитации, а также в нормальной жизнедеятельности и адекватном восприятии
их и их возможностей окружающим народом.
Российский филиал функционирует в режиме поддержки родителей детей, пораженных этой болезнью, в консультации и оказании медицинской помощи.
В 2020 году в Казани прошел конгресс, в котором приняли участие доктора таких стран, как Израиль, Россия, Япония, Италия, Нидерланды и США.
По их мнению, синдром Ретта, что поражает столь большое количество детей, остается незаметным для глаз простых педиатров. В дальнейшем же, после того, как недуг проявит себя в полной мере, у ребенка нет возможности развиваться в реабилитационных центрах (которых попросту нет) или лечиться. Поэтому было принято решение создать ассоциацию
, которая смогла бы помочь всем нуждающимся в данном деле.
Лечение
Синдром Ретта — генетическое заболевание, которое абсолютно не поддается терапии. Современная медицина бессильна перед генетикой. Симптоматическое лечение, облегчающее жизнь больным, также сопряжено с рядом трудностей. Синдром Ретта в настоящее время лечат в специализированных реабилитационных центрах, которые имеются практически во всех крупных городах нашей страны. Специалисты центров проводят развивающие занятия, адаптируя маленьких пациентов к окружающему миру.
Медикаментозная терапия смягчает симптоматические проявления патологии и облегчает общее состояние. Больным назначают:
- Ноотропные препараты, улучшающие мозговую микроциркуляцию и стимулирующие работу органа – «Пирацетам», «Винпоцетин», «Цинноризин», «Мексидол», однако существенного эффекта от их применения не отмечено,
- Снотворные средства на основе мелатонина – «Мелаксен», «Циркадин», «Тразодон»,
- Антиконвульсанты, блокирующие приступы эпилепсии – «Карбамазепин», «Вальпарин»,
- Стимуляторы дофаминовых рецепторов – «Бромокриптин», «Толкапон», «Леводопа»,
- Психотропные препараты – «Фенибут», «Глицин», «Ноофен»,
- Препараты для лечения заболеваний сердца, печени, селезенки.
Лечебный рацион показан с целью предупреждения истощения больных. Им рекомендовано частое и дробное питание, употребление продуктов с высоким содержанием жиров, клетчатки и витаминов. Диету составляют индивидуально каждому больному для набора веса.
Физиотерапевтические процедуры, лечебный массаж, ЛФК — обязательные компоненты сложного лечебного процесса. Такие занятия способствуют развитию конечностей ребенка, повышают их гибкость, стимулируют мышечный тонус. Массаж и гимнастика поддерживают в исправном состоянии опорно-двигательный аппарат и противостоят дальнейшей деградации.
Лечение музыкой активно применяется при синдроме Ретта. Такая терапия благотворно воздействует на организм, успокаивает и стимулирует интерес к окружающему миру. Работа с дефектологом, психологом, остеопатом помогает ребенку улучшить навыки коммуникации и моторику. Остеопатия положительно влияет на состояние позвоночника. Мануальная терапия обычно сочетается с другими методами реабилитации. Хороший эффект дают занятия со специально обученными животными, гидрореабилитация, арт-терапия.
В настоящее время проводятся генетические и фармакологические исследования различных групп препаратов для лечения синдрома Ретта.
Симптомы синдрома Ретта
В перинатальный период развития плода и в первые месяцы жизни ребенка какие-либо симптомы заболевания не проявляются. Возможно, они просто остаются недооцененными специалистами, но у врачей подозрений не вызывают. Тем не менее, достаточно часто у детей с синдромом Ретта наблюдается врожденная гипотония, однако далеко не у всех детей с гипотонией имеется эта аномалия.
Условно течение заболевания делят на четыре стадии, каждая из которых характеризуется своей симптоматикой.
1. Первая стадия (стагнация) характерна для малышей в возрасте от 4-х месяцев до 2-х лет. В этот период их жизни можно наблюдать первые отклонения, проявляются они замедлением роста головы, в длину кистей и стоп, а также диффузной мышечной гипотонией, медленным психомоторным развитием, исчезновением интереса к играм.
2. Вторая стадия (быстрого регресса) характерна для детей в возрасте от 1-го до 3-х лет. Проявляется нервно-психическим регрессом, смягчающимися аутистическими расстройствами, общим моторным беспокойством, апраксией, быстро наступающим распадом речи. Постепенно, но достаточно быстро у ребенка пропадают все навыки (вплоть до умения разговаривать), которые он успел приобрести до этого времени. Параллельно появляются характерные движения руками – моющего или потирающего характера. Дети жуют пальцы рук, царапают себя, могут наносить удары кулаками по щекам. Все эти движения совершаются насильственно и непрерывно (реже с небольшими интервалами). У больного пропадает способность удерживать предметы в руках, меняется походка – он при ходьбе широко расставляет ноги и двигается покачиваясь (так называемая атактическая походка). У многих детей развивается апноэ, чередующийся с гипервентиляцией, иногда появляются судорожные припадки. Основной симптом синдрома Ретта этой стадии – потеря общения с окружающими, из-за чего это заболевание часто воспринимают за аутизм.
3. Третья стадия (псевдостационарная) характерна для детей дошкольного и раннего школьного возраста. Первые признаки этого периода – глубокая умственная отсталость, приступы судорог и экстрапирамидные расстройства, такие как атаксия, гиперкинезы и мышечная дистония. Но отмечается и положительная картина – у ребенка прекращаются приступы беспокойства, улучшается сон, появляется эмоциональный контакт.
4. Четвертая стадия (тотального слабоумия) развивается примерно к 10 годам. Для нее характерны нарушения всех двигательных функций, вплоть до обездвижения больного, а также нарастание мышечной атрофии, спастичности, вторичных ортопедических деформаций в виде сколиоза, вазомоторных расстройств нижних конечностей. Дети начинают отставать в росте, у них развивается кахексия (глубокое истощение). Однако реже бывают приступы судорог, и половое созревание соответствует возрасту.
Синдром Ретта характеризуется прогредиентным течением, т.е. постепенно нарастающими изменениями. Определенных границ между описанными стадиями не существует, они взяты условно.
Типичные и наиболее важные клинические признаки этой аномалии:
- Атаксия:
- Эпилептические припадки;
- Вторичная микроцефалия;
- Низкий рост, маленькие руки;
- Судороги мышц;
- Потеря ловкости рук;
- Стереотипные движения рук (будто ребенок их моет или потирает);
- Скрежет зубами;
- Боковое искривление позвоночника;
- Проблемы с дыханием;
- Избегание зрительного контакта;
- Утрата способности говорить;
- Приступы паники;
- Связанные с социальными контактами проблемы.
Диагностика заболевания
Диагноз ставит психиатр на основании опроса родителей и характерной клинической картины заболевания.
Международной ассоциацией по исследованию патологии были предложены диагностические критерии, разделённые на три группы: обязательные, второстепенные и исключающие.
К первой группе критериев относятся симптомы, которые могут свидетельствовать о классической форме синдрома. Обязательно наличие следующих признаков:
- беременность протекала нормально, в родах и послеродовом периоде осложнений не было;
- развитие до 5–18 месяцев было по возрасту;
- окружность головы при рождении соответствовала норме, замедление её роста началось в период от 5 месяцев до 4 лет;
- нарушение произвольных движений рук в период с полугода до двух с половиной лет;
- сильная задержка психомоторного развития, грубое нарушение речи;
- характерные стереотипии;
- расстройство координации движений, походки в возрасте 1–4 лет.
Дополнительные (второстепенные) признаки:
- нарушения дыхания;
- судорожные припадки;
- искривление позвоночника;
- задержка роста, в том числе гипотрофия ступней и кистей.
Исключающие критерии позволяют дифференцировать синдром Ретта с другими неврологическими патологиями. К ним относятся:
- внутриутробная задержка роста и другие аномалии развития;
- врождённая микроцефалия;
- перинатальное повреждение головного мозга;
- черепно-мозговые травмы;
- нейроинфекции.
Если есть хотя бы один такой признак, синдром Ретта исключается. Иногда набор симптомов не совсем соответствует классическому течению патологии. Тогда заболевание расценивают как неполную или атипичную форму.
Диагностические мероприятия начинаются с осмотра ребёнка психиатром и опроса родителей
Опросив родителей и осмотрев ребёнка, психиатр назначает дополнительные обследования:
- МРТ — позволяет выявить атрофию мозга лёгкой или средней степени, иногда атрофию мозжечка
- ЭЭГ (электроэнцефалограмма) регистрирует прогрессирующее замедление фоновой активности, изменения, характерные для эпилепсии.
Электроэнцефалограмма является дополнительным методом обследования ребёнка с синдромом Ретта
- Рентгенография стоп и кистей у большинства больных показывает укорочение локтевой, четвёртой плюсневой и пястной костей.
- УЗИ внутренних органов у четвёртой части больных показывает пороки развития селезёнки и печени.
- Методы современной генетики дают максимально полную диагностическую информацию, позволяя выявить мутацию гена, ответственного за развитие заболевания.
Дифференциальная диагностика
Дифференциальная диагностика главным образом проводится с детским нейрональным цероидным липофусцинозом (НЦЛ), детским церебральным параличом (ДЦП) и ранним аутизмом.
Для НЦЛ характерен стремительный регресс двигательных и коммуникативных навыков, этот диагноз исключается посредством проведения тканевого исследования электронным микроскопом.
Симптоматика синдрома Ретта очень сходна на раннем этапе с проявлениями аутизма:
- асоциальность ребёнка (нежелание контактировать с внешним миром и окружающими);
- нарушение речи;
- снижение способности к обучению; недержание кала и мочи;
- беспричинное беспокойство;
- повторяющиеся движения.
Таблица — дифференциальная диагностика с аутизмом
| Признаки | Синдром Ретта | Аутизм |
| Нормальное развитие до 6–18 месяцев | типично | не типично |
| Стереотипии | характерные движения руками | могут быть задействованы все части тела, движения разнообразные и сложные |
| Повторяющиеся манипуляции с различными предметами | не типично | повторяются постоянно |
| Моторика, координация движений, походка | прогрессирует апраксия и атаксия вплоть до полной неподвижности | нарушение общей и тонкой моторики, недостаточно координированная походка |
| Судорожный синдром | типичный признак | проявляется редко |
| Дыхательные нарушения | типичный симптом | не наблюдаются |
| Микроцефалия, гипотрофия стоп и кистей | всегда присутствуют | не типичны |
Что такое «синдром Ретта»?
Синдром Ретта — прогрессирующее дегенеративное заболевание. Оно оказывает поражающее воздействие на центральную нервную систему. В частности, патология подвергает изменениям серое вещество головного мозга.
Впервые патологию выявили в 1954 году. Андреас Ретт, работавший педиатром, заметил у двух девочек стереотипные движения рук и нарушение психического развития. К 1983 году было выявлено ещё 30 случаев с аналогичной симптоматикой. Это позволило патологии получить статус самостоятельного заболевания.
Статистика показывает, что нарушение появляется один раз на 10000-15000 здоровых детей. Чаще с заболеванием сталкиваются девочки. Однако развитие симптоматики возможно и у мальчиков. Патология не зависит от расовой и этнической принадлежности.
Патология проявляется серией неврологических нарушений. Они выражаются в аутичном поведении и проблемами с мелкой и крупной моторикой. У пациентов с заболеванием наблюдается остановка роста головы, рук и ног. Дополнительно присутствуют расстройства ЖКТ и сколиоз. Статистика показывает, что около 80% детей, у которых выявлена патология, страдают эпилептическими припадками. Порядка 50% пациентов самостоятельно передвигаться не могут.
До 1-1,5 лет дети развиваются вполне нормально. Затем начинает проявляться симптоматика заболевания. Своим поведением дети очень напоминают больных ранним аутизмом. В некоторых случаях возможно более раннее возникновение заболевания. Самые первые признаки удаётся заметить в возрасте 4-6 месяцев. Они проявляются задержкой в развитии. Однако диагностировать недуг на этом этапе достаточно сложно. В последующем заболевание начинает прогрессировать, приобретая характерные особенности. Всё это упрощает постановку диагноза. Синдром Ретта является генетической патологией. Всё это приводит к тому, что во взрослом возрасте приобрести заболевание невозможно. Патологии диагностируются исключительно у детей.
Повышение риска возникновения заболевания у девочек происходит из-за того, что ген, в котором происходит мутация, находится в X хромосоме. У женщин их две. Если у младенца женского пола имеется один нормальный ген, а другой мутировавшей, это повышает жизнеспособность ребёнка. Плод мужского пола обладает только 1 x-хромосомой. Если в ней происходит мутация, он погибает еще в утробе. Однако случаи рождения ребенка мужского пола с синдромом Ретта всё же существует. В этом случае патология часто сопровождается синдромом Клайнфельтера. Дело в том, что такие дети обычно являются носителями лишней x-хромосомы.
В течение последних 15-20 лет ученые начали активно изучать заболевание. Реальные исследования практически не проводились. До сих пор точные причины возникновения патологии не выявлены. Однако у врачей имеется повод для того, чтобы считать, что заболевание передаётся по наследству. Это удалось доказать благодаря проведению территориального анализа распространения заболевания. В ходе исследования, которые прошли в Албании, Италии, Венгрии и Норвегии, удалось найти несколько небольших поселений, в которых больные синдромом Ретта встречались чаще всего.
Причины появления
В основе развития патологии лежит мутация гена MECP2, который расположен в X-хромосоме.
Его экспрессия прерывается, и он прекращает функционировать корректно.
Если этот ген в норме, он в определенный период формирования мозга блокирует активность ряда других генов, и мозг продолжает полноценное развитие.
Если структура гена нарушена, он не выполняет эту функцию, и мозг формируется некорректно, что приводит к возникновению прогрессирующей умственной отсталости и прочих тяжелых симптомов.
Более 70% случаев возникновения патологии связаны с нарушениями в генетическом материале отца ребенка.
Как обнаружить и подтвердить диагноз?
При подозрении на болезнь Ретта следует обратиться к психотерапевту. Он на основе опроса о наблюдениях родителей, течения беременности и жалоб, а также проведя осмотр ребенка, может подтвердить или опровергнуть подобные догадки. В любом случае диагноз требует дополнительного подтверждения. С этой целью пациента направляют на обследование по таким направлениям:
- Компьютерная томография мозга. Показывает затухание активности ЦНС путем оценки изменений.
- Электроэнцефалограмма. Отмечает биоэлектрическую активность мозга. Характер показаний может напрямую свидетельствовать о наличии генной патологии Ретта.
- Генетический анализ крови на подтверждение поломки X-хромосомы.
- Ультразвук всех внутренних органов. Для выявления осложнений синдрома.
Ребенку проводят комплексную диагностику состояния с привлечением большинства профильных специалистов, чтобы понять, какие патологии развились в результате синдрома.
В период планирования зачатия мать может сделать генетический анализ на кариотип, чтобы исключить носительство данной мутации. Однако последующую поломку хромосомы трудно предугадать. Анализ околоплодных вод, крови из пуповины или кусочка плаценты не делают, так как данная патология не диагностируется во время беременности, плод развивается нормально. Кроме случаев с мальчиками, тогда аномалии проявляются уже при внутриутробном периоде.
В чем отличие от аутизма?
Порой болезнь Ретта путают с аутизмом. Разграничить эти состояния может только специалист. В 1988 году был разработан метод дифференциальной диагностики для постановки точного диагноза.
Можно отметить ряд внешних и поведенческих отличий:
- Особые движения рук при синдроме ограничиваются их положением. Как правило, они согнуты в локтях, кисти чуть приподняты, находятся у груди. При аутизме движения более разнообразны и не сосредоточены в одном месте.
- Дети с болезнью Ретта до 1,5–2 лет способны к общению, реагируют на внешний мир, тогда как малыши-аутисты уже в этом возрасте испытывают проблемы.
- При синдроме серьезно страдает возможность ходить, тогда как при аутизме это лишь некоторая характерность походки.
- Сила и частота судорожных приступов выше при патологии Ретта.
- При аутизме отсутствует бруксизм, угнетение роста, нарушения дыхательной системы.
Стадии болезни
Выделяют 4 стадии заболевания:
- Первая стадия развивается в начале четвертого месяца жизни и может продолжаться до двух лет. Появляются отклонения физического развития ребенка, отсутствует интерес к происходящему, апатия, задержка роста головы, набора веса, низкий мышечный тонус и вялость.
- Вторая стадия развивается к году жизни, когда ребенок ползает, переворачивается, ходит, говорит. В течение года эти приобретенные навыки исчезают. Он не помнит, как разговаривать, ходить и т.д. У ребенка возникают стереотипные движения рук, напоминающие мытье под краном, нарушение дыхания во время отдыха и не только, расстройство координации движений. Также возникает судорожный синдром, напоминающий эпилептический припадок. Лечение эпилептического припадка не приводит к исчезновению судорог.
- Третья стадия чаще наблюдается в конце второго начале третьего годика. Этот этап еще называют стабилизацией. На этом этапе происходит регресс развития головного мозга, т.е развивается умственная отсталость, уменьшается количество эпилептиморфных припадков. Ребенок может находится в одном положении длительное время (находится «в себе»), но при этом нет беспокойства и криков.
- Четвертая стадия является самой тяжелой, развивается примерно к 5 -10 годам и характеризуется необратимыми дегенеративными процессами в центральной и периферической нервной системах, а также в суставах и позвоночном столбе. Развивается искривление позвоночника, ведущее в 3-4 степени сколиоза. Характерна кахексия (снижение веса ребенка), мышечная гипотония, уменьшение размеров головного мозга, боли в суставах и мышцах. Эпилептические припадки появляются очень редко.
Течение заболевания основывается на скорейшем установлении диагноза. Чем раньше был установлен диагноз, проведены лечебные и профилактические мероприятия, тем благоприятнее течение болезни. У разных детей течение патологии происходит по-разному. Если установление диагноза было выставлено в поздние сроки, велик риск развития тяжелейших осложнений, препятствующих нормальной жизни больного ребенка.
Симптомы синдрома Аарскога
Синдром Аарскога прежде всего поражает мужчин. У пораженных мальчиков наблюдается характерный набор лицевых, скелетных и генитальных аномалий. Клинические признаки могут варьироваться от человека к человеку (клиническая неоднородность), даже в семьях.
Мужчины с синдромом Аарскога часто имеют округлое лицо с широким лбом (см. фото).
Дополнительные характерные черты лица включают:
- широко расставленные глаза (глазной гипертелоризм);
- опущение век;
- наклоненные вниз складки век;
- маленький нос с ноздрями, которые расширены вперед;
- недоразвитая верхняя челюсть;
- мыс вдовы (особенность роста волос на лбу).
Пострадавшие могут также иметь:
- аномально длинную бороздку на верхней губе (филтрум);
- широкий носовой мостик.
Больные дети также могут иметь различные нарушения, влияющие на уши и зубы.
Ушные аномалии включают:
- низко посаженные уши;
- утолщенные, «мясистые» мочки уха.
Зубные аномалии включают:
- отсутствие зубов при рождении;
- отложенное прорезывание зубов;
- недоразвитие твердого наружного покрытия зубов (гипоплазия эмали).
Синдром Аарскога в основном представляет собой дисплазию скелета, и у пораженных мужчин развиваются характерные пороки развития скелетной системы, включая:
- непропорционально низкий рост;
- широкие, короткие руки и ноги;
- короткие пальцы (брахидактилия) с постоянной фиксацией пятого пальца в согнутом положении (клинодактилия);
- ненормально растяжимые суставы пальцев;
- широкие плоские ноги с выпуклыми пальцами.
Кроме того, у пораженных людей может быть:
- утопленная грудная клетка;
- выпячивание участков толстой кишки через аномальное отверстие в мышечной оболочке брюшной полости (паховая грыжа);
- выступающий пупок.
Далее у людей с синдромом Аарскога могут быть такие нарушения позвоночника, как:
- незаращение дужки позвонка (расщепление позвоночника);
- слияние верхних костей позвоночника;
- недоразвитость второго шейного позвонка (одонтогенная гипоплазия).
Признаками, которые помогают поставить диагноз у мужчин с синдромом Аарскога, являются генитальные аномалии, включая:
- характерную аномальную складку кожи, распространяющуюся вокруг основания полового члена;
- неспособность одного или обоих яичек опускаться в мошонку (крипторхизм);
- отверстие для мочеиспускания может быть расположено на нижней стороне полового члена (гипоспадия);
- мошонка может быть расщеплена или разделена (раздвоенная мошонка).
У некоторых пострадавших мальчиков была описана интеллектуальная неполноценность, но она не является постоянной чертой этого расстройства. Пострадавшие люди могут иметь ряд легких трудностей в обучении и/или поведенческих нарушений:
- у пострадавших детей может наблюдаться задержка развития в младенческом возрасте;
- гиперактивность;
- дефицит внимания;
- импульсивность;
- обструкционность.
Также описаны неспособности набирать вес, расти с ожидаемой скоростью и развитие хронических респираторных инфекций.
Дополнительный спектр признаков и симптомов, встречающихся реже, включают:
- врожденные пороки сердца;
- эпилепсия;
- аномальная поперечная кривизна позвоночника (сколиоз);
- дополнительные пары ребер;
- расщепление нёба (волчья пасть)
- вертикальная бороздка в верхней губе (заячья губа);
- мягкая перепонка пальцев;
- короткая шея с или без лямки.
Могут присутствовать дополнительные аномалии глаза, включая:
- перекрещенные глаза (косоглазие);
- дальнозоркость (гиперметропия);
- паралич определенных глазных мышц (офтальмоплегия).